Dashboard
Modern applications call for solutions that handle "big data," datasets that span multiple machines and cannot fit in main memory, and "big compute," computation patterns that tax even the most advanced processors. Bioinformatics is no exception.
We present HBaaS, Heterogeneous-accelerated Bioinformatics-as-a-Service. Our platform leverages heterogeneous computer architectures to provide sequence matching and motif finding as a web service for users of bioinformatics data. We tackle big data by means of the Accumulo distributed database, and big compute by means of GPUs. Their integration delivers top-tier performance for our chosen applications..
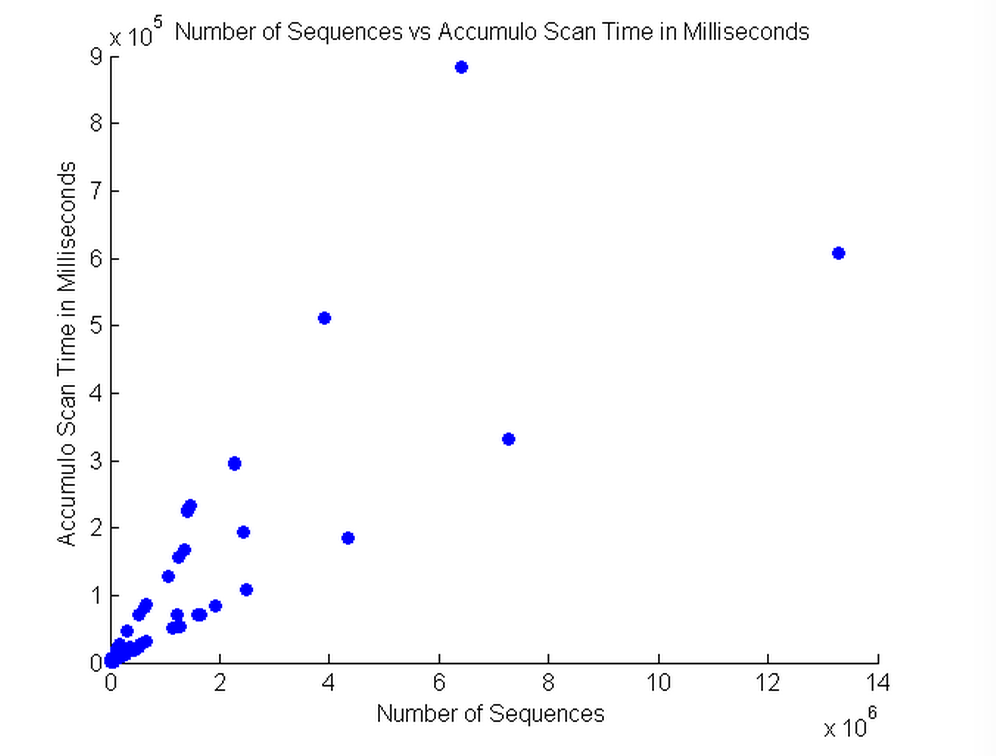
This graph shows us the number of sequences vs time it takes to scan. We can see two distinct lines which may be an artifact of how the data was ingested into accumulo. It takes roughly 10 minutes to scan 14 million sequences based on the line with a smaller slope. This is a very naive graph because we are using a small batch size of 10,000 and only have 1 thread. This is not using the full power of the GPUs. However, it shows that Accumulo is handling the data well because we are seeing data in a linear pattern instead of an exponential curve.
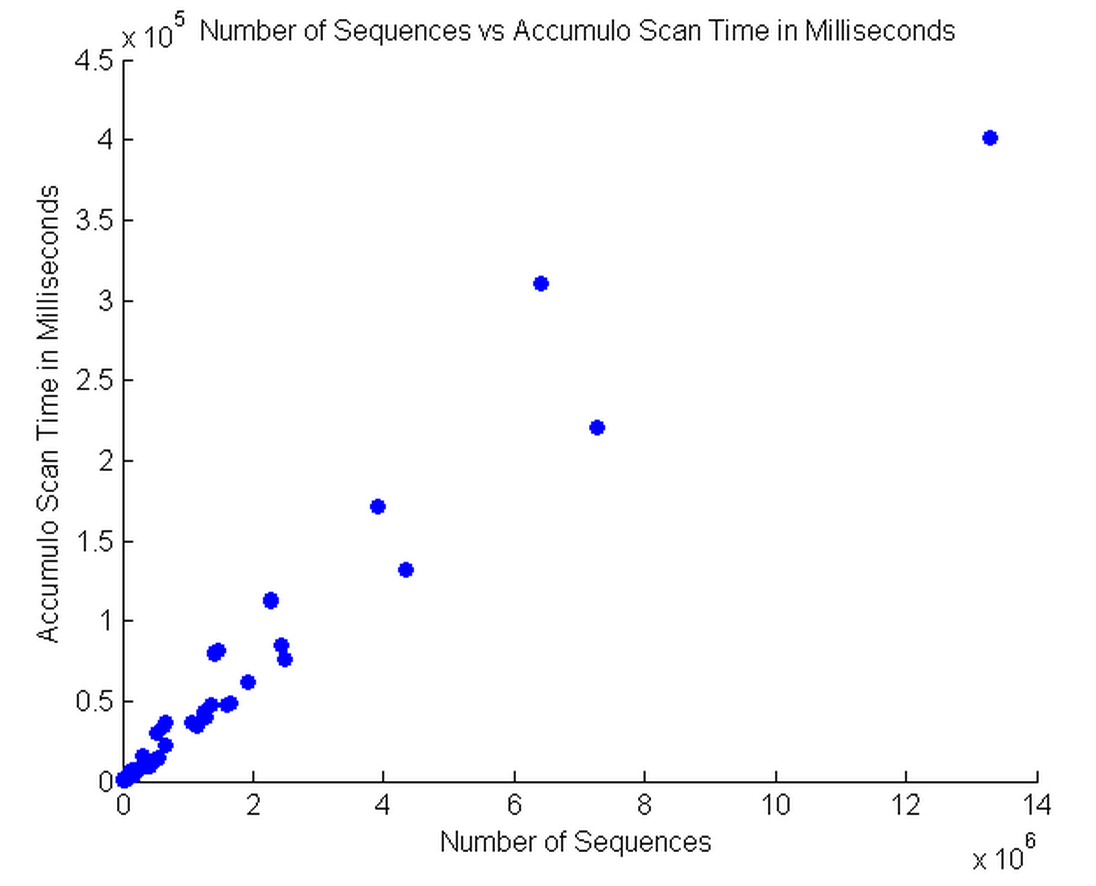
This graph shows the number of sequences vs time it takes to scan. We can see two distinct lines are converging. We used 4 threads with a batch size of 10,000 which resulted in a speedup of roughly two times to scan the database. We see that it only takes about 7.5 minutes to scan 14 million sequences. We plan on improving performance by installing Accumulo onto other nodes in order to further decrease scan times.
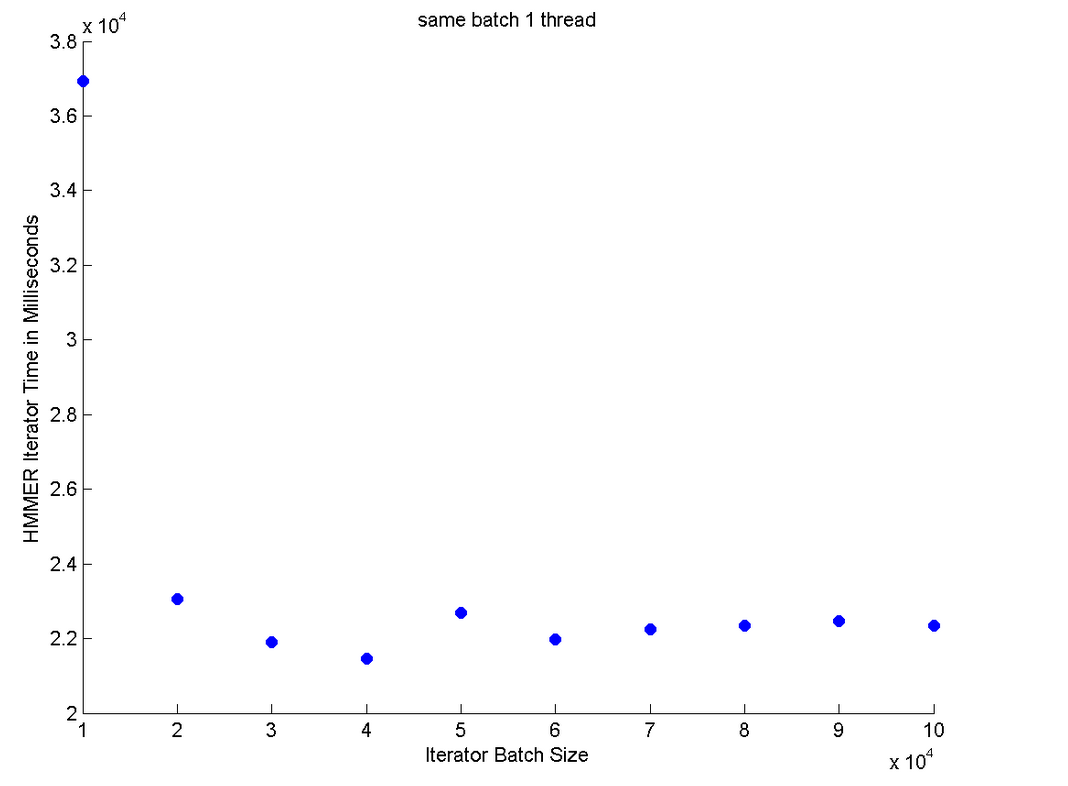
This is our worst performance graph. It uses only a single thread for the iterator. The time it takes for the iterator appears to be independent of the batchsize of a single thread.
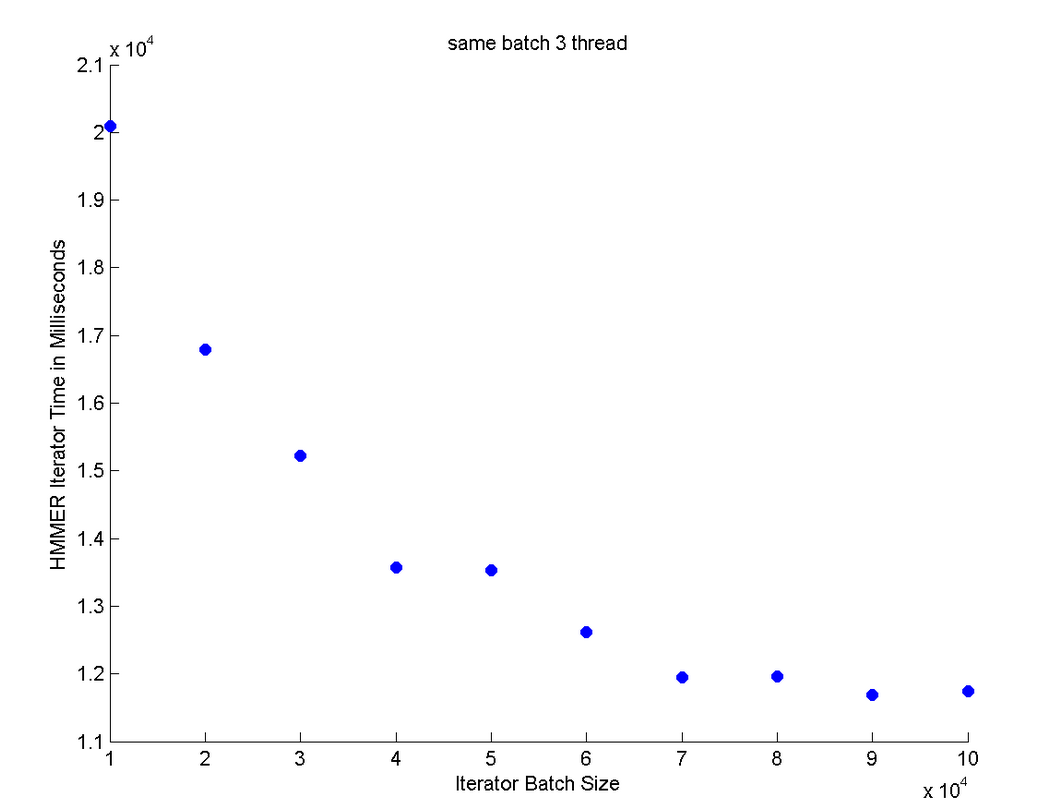
This graph was the best performance graph because it took much less time to process the data. With a thread size of 3 and a batch size of 100,000, we were able to process the data in about 12 seconds. The graph shows an exponential decrease that levels off when we increase the batch size.
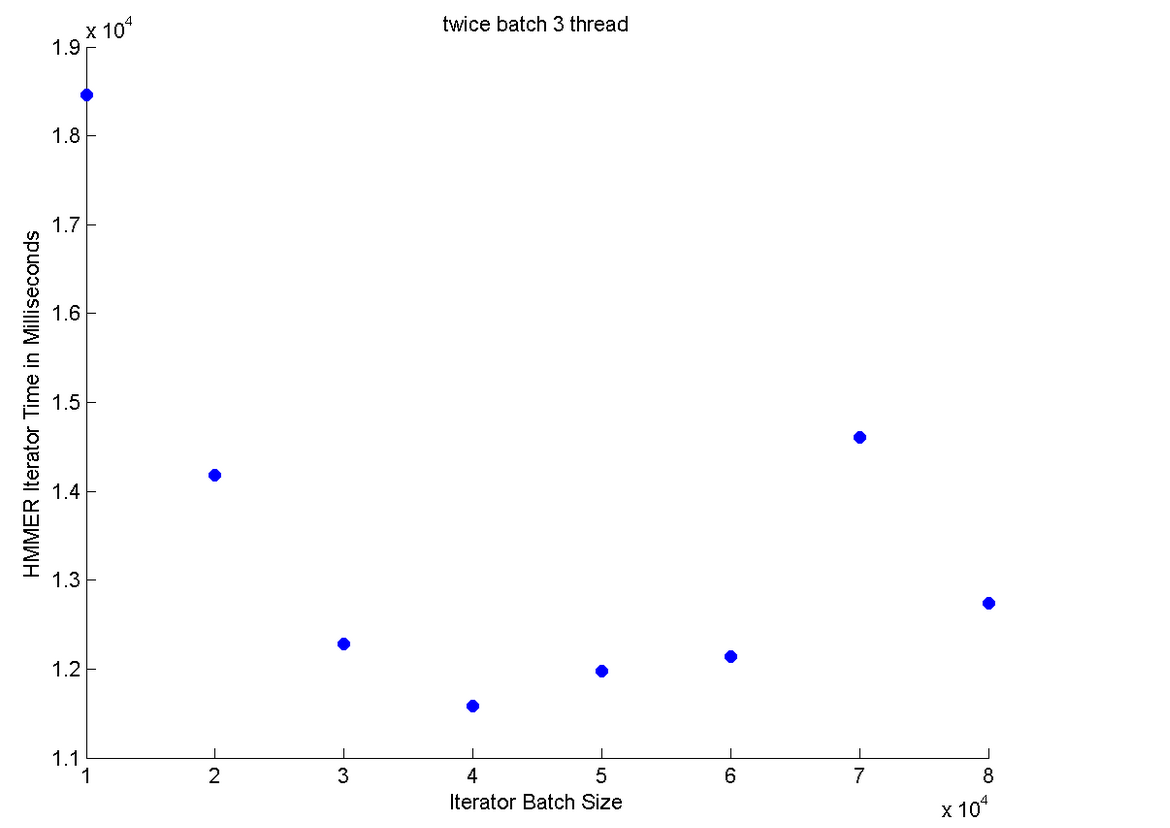
We also tried to prescan the tables to collect twice the amount of sequences that the batch iterator required. We reasoned this would speed the iterator time. However, we are memory restricted so holding more data will be a problem. We abandoned this approach because the speed up times are not much better.